馬凡氏綜合症
馬凡氏綜合徵又叫馬凡綜合徵,是一種臨床患者表現為體格細高、四肢及指(趾)細長,特別是四肢、手指、腳趾細長不勻稱等體徵的先天遺傳性結締組織疾病。一般經過檢查和詢問病史、家族史,並經彩色B超和磁共振檢查後即可確診馬凡氏綜合徵,而一旦確診的話就要及時進行治療,這樣才能避免心力衰竭、主動脈破裂和心肌梗塞的發生。
目錄
- 1 馬凡氏綜合症是什麼
- 2 馬凡氏綜合症的症狀有哪些
- 3 “天才病”馬凡氏綜合徵的診斷方法
- ▪ 骨骼系統
- ▪ 眼睛系統
- ▪ 心血管系統
- ▪ 其他
- ▪ 馬凡綜合徵的診斷前提
- 4 馬凡氏綜合症患者真人照片
- 5 馬方綜合徵治療方法
- ▪ 一般性藥物治療
- ▪ 手術治療
- 6 相關疑問
馬凡氏綜合症
基本資訊
- 中文名
- 馬方綜合徵
- 別名
- 馬凡綜合徵等
- 常見症狀
- 身材高大、蜘蛛指
- 外文名
- Marfan syndrome
- 常見病因
- 常染色體顯性遺傳
- 就診科室
- 心血管外科
馬凡氏綜合症是什麼
馬凡氏綜合徵又叫馬凡綜合徵、先天性中胚層發育不良,它是一種比較少見的先天遺傳性結締組織疾病,造成的原因為常染色體顯性遺傳,伴隨有家族史。
| 病症名稱 | 馬凡氏綜合徵 | 英文名稱 | Marfan syndrome |
| 別稱 | 馬凡綜合徵、先天性中胚層發育不良、蜘蛛指徵、肢體細長症 | ||
| 就診科室 | 心血管外科 | 型別 | 遺傳性結締組織疾病 |
| 致病原因 | 常染色體顯性遺傳,原纖維蛋白-1基因是最常見致病基因 | ||
| 臨床症狀 | 身材高大、蜘蛛指、先天性心血管畸形、晶狀體異位 | ||
| 患病明星 | 霍萱、朱剛、韓朋山、張佳迪、武強等 | ||
雖然馬凡氏綜合徵能夠帶給患者異於常人的運動體格,但患者往往伴隨有心血管系統異常,特別是合併的心臟瓣膜異常和主動脈瘤,不注意情況下會導致猝死,可謂體內的“不定時炸彈”。
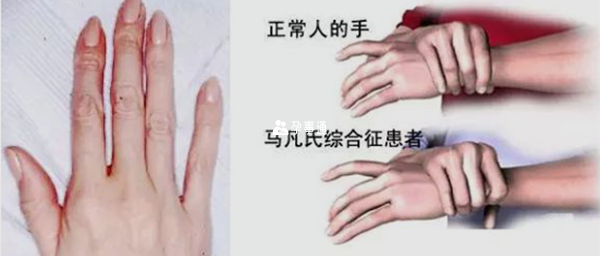
患有馬凡氏綜合症的患者常表現的症狀為體格細高、四肢及指(趾)細長,特別是四肢、手指、腳趾細長不勻稱。95%的馬方綜合徵患者死於心血管併發症,常見為動脈瘤破裂和充血性心力衰竭。未經治癒的患者平均壽命為男性30歲左右,女性40歲左右。
馬凡氏綜合徵的發病率約為0.04‰~0.1‰,據推測中國大約有16萬人患有此病。要知道馬方綜合徵雖然不能根治,但越早發現對於後期預後有著很大的作用,同時也能延長患者生命。
馬凡氏綜合症的症狀有哪些
馬凡氏綜合症是由於人類第15號染色體上的原纖維蛋白基因(Fibrillin-1,FBN1)缺陷所致。原纖維蛋白單體是由42個氨基酸所組成的多肽,是彈性纖維的基本成分,它也完全依賴於鈣和半胱氨酸二硫鍵的結合。
當馬凡氏綜合症時,半胱氨酸被其它氨基酸所替代,不能與鈣結合,導致彈性纖維變性、細小和退變,從而引起動脈層中的囊性壞死和斷裂,病理上可發現III-IV級中層退行性變。
關於馬凡綜合徵的症狀常見的表現為患者多體型頎長,柔軟度超人。有資料顯示在兒童時期馬凡氏綜合徵患者39%無心血管病變,僅有身高增長速度過快而體重增長相對遲緩。
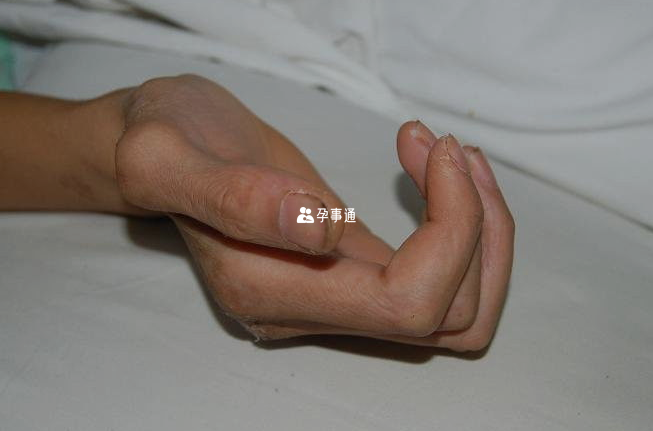
兒童首次診斷為馬凡氏綜合症大多是根據心血管以外的一些症狀表現,比如75%是近視(晶狀體半脫位),83%是脊檢側彎或者蜘蛛指(趾)等其他畸形。
成年馬凡氏綜合徵患者的話,除了體格異常與關節鬆弛等症狀外,主要是心臟功能不全及由於升主動脈瘤壓迫出現的症狀,如心悸、胸悶、氣短、胸痛等。
“天才病”馬凡氏綜合徵的診斷方法
馬凡氏綜合徵的診斷標準最早在1987年第七屆人類遺傳學國際大會上建立,並於1988年第一次國際馬凡氏綜合徵專題討論上明確規定,隨後1996年de Paepe A重新修正被視為統一標準。
骨骼系統
針對骨骼系統的診斷標準分為主要和次要,是否為馬凡綜合徵患者,需符合的條件包括至少兩項主要標準或一項主要標準加兩項次要標準。
骨骼系統症狀
1、雞胸;
2、漏斗胸需外科矯治;
3、上部量/下部量的比例減少,或上肢跨長/身高的比值大於 1.05;
4、腕徵、指徵陽性;
5、脊柱側彎大於 20 度,或脊柱前移(側彎計);
6、肘關節外展減小(〈170度);
7、中踝中部關節脫位形成平足;
8、任何程度的,髖臼前凸(髂關節內陷)(X 片上確定)。
次要標準:中等程度的漏斗胸:關節活動異常增強;高顎弓,牙齒擁擠重疊;面部表徵:長頭——正常頭顱指數為 75.9 或以下、顴骨發育不全、眼球內陷、縮頜、瞼裂下斜。
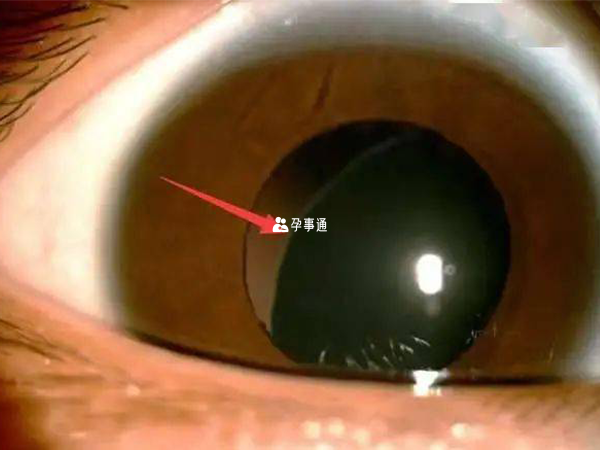
眼睛系統
主要標準是晶狀體脫位,次要標準包括異常扁平角膜(角膜曲面計測量);眼球軸長增加(超聲測量);虹膜或睫狀肌發育不全致瞳孔縮小。眼睛系統受累需符合標準:主要標準或至少兩項次要標準。
心血管系統
心血管受累需符合的條件:有一項主要標準或一項次要標準即可。
次要標準
1、二尖瓣脫垂伴或不伴二尖瓣返流;
2、主肺動脈擴張( 在無瓣膜或外周肺動脈狹窄及其它明顯原因下,年齡又小於 40 歲);
3、二尖瓣環鈣化(年齡小於 40 歲);
4、降主動脈或腹主動脈擴張或夾層(50 歲以下)。
主要標準:升主動脈擴張伴或不伴主動脈瓣返流,以及至少 Valsava 氏竇擴張;升主動脈夾層。
其他
其他包括肺系統、面板和體包膜、家族或遺傳史等,具體如下:
肺系統
- 自發性氣胸;肺尖肺大泡(胸片證實)。
面板和體包膜
- 皮紋萎縮(牽拉痕),與明顯超重、妊娠或反覆受壓等無關;複發性疝或切口疝。一項次要標準存在即可認為面板或體包膜受累。(次要標準)
家族或遺傳史
- 父母、子女或兄弟姊妹之一符合該診斷標準;FBNI 基因中存在已知的導致馬凡綜合徵的突變;存在已知的與其家族中馬凡綜合徵患者相同的 FBNI 基因單倍型。(主要標準)
注:由於家族或遺傳史在診斷中意義重大,主要標準中必須有一項存在。
馬凡綜合徵的診斷前提
對特定病例:如果無家族或遺傳史者,至少需有兩個不同系統的主要標準以及三分之一的器官受累;如果檢出一個已知馬凡綜合徵的基因突變,一個系統中有一項主要標準和第二項系統受累即可診斷。
需與馬凡綜合徵鑑別的疾病
1、先天性攣縮性蜘蛛指徵(Viljoen,et al.1994);
2、家族性胸主動脈瘤(Savunen 1987;Emanuel,et al. 1977), 過去稱為埃德海姆囊性中層壞死;
3、家族性主動脈夾層(Nicod,et al. 1989);
4、家族性晶狀體脫位(Tsipouras,et al.1992);
5、家族性類馬凡體型(Milewicz,et al.1995);
6、家族性二尖瓣脫垂綜合徵(Devereux,et al.1982,1986,1987 ; Roman ,1989b);
7、Shprintzen-Goldberg 綜合徵(Shprintzen和 Goldberg,1982);
對特定病例的家屬:在家族史中有一項主要標準、一個系統的一項主要標準和第二個系統受累即可診斷。
關於馬凡氏綜合症新診斷標準與1988年的診斷標準的主要區別在於,對家族中無典型馬凡綜合徵患者的診斷要求更為嚴格,同時骨骼作為一項主要標準,8 項表現中必須至少有4項方可考慮骨骼受累等。
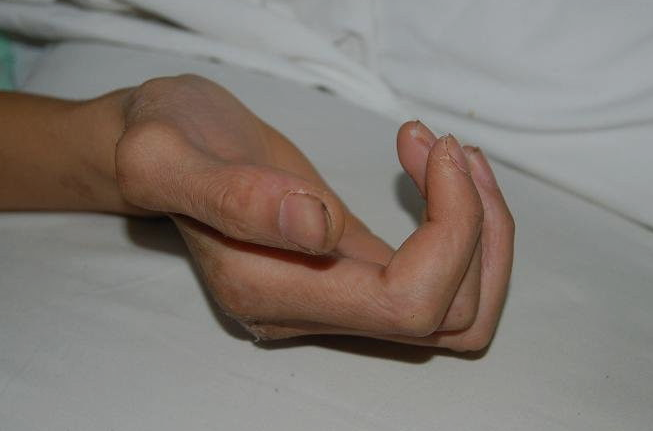
馬凡氏綜合症患者真人照片
因馬凡氏綜合徵發生的悲劇不少,早期中國女排前國手霍萱、美國女排名將海曼、四川男排國手朱剛以及男籃運動員張佳迪和武強均因此病猝死,“馬凡氏”已奪走多名運動員生命,以下是部分馬方綜合徵患者真人照片:


馬凡氏綜合徵除極少數是基因突變而來外,大部分是遺傳產生的。病人多患有心血管系統異常,一旦出現主動脈異常隨時會危及生命,如果不及時手術,存活的希望幾乎為零。
馬方綜合徵治療方法
其實現實中針對馬凡氏綜合徵的診斷並非難事,一般經過檢查和詢問病史、家族史,並經彩色B超和磁共振檢查後即可確診。而一旦確診的話就要根據情況進行手術治療,只有早發現早治療,才能避免心力衰竭、主動脈破裂和心肌梗塞的發生。
一般性藥物治療
針對馬凡氏綜合徵的治療方法一般建議手術為主,因為目前來說藥物治療或其他保守療法均不能去除此病。藥物治療的主要目的是減緩或推遲心血管病變的發生,防止心率失常。
手術治療
隨著科技進步,目前馬凡氏綜合徵手術成功率已在90%以上。當馬凡氏綜合徵有出現心血管病變時就只能通過手術治療。一旦出現心血管病變,就必須馬上治療,否則預後極差。
手術治療適應症
1、主動脈根部徑>50mm;
2、有家族史(猝死或主動脈夾層)主動脈根部徑≥45mm;
3、主動脈根部增長率>5mm/年;
4、兒童根部瘤直徑擴張大於其正常直徑50%應手術治療。
可能有人會擔心馬凡氏綜合徵的治療費用昂貴負擔不起,實際上馬凡氏綜合徵治療費用與做主動脈瘤的手術費用差不多,都是在15萬左右。如果是發現早、治療早,病變沒有出現主動脈夾層,或者說沒有廣泛病變,這種情況下治療徹底且費用較低。
相關疑問
Q:馬凡氏綜合徵是否為禁止結婚的遺傳疾病?
A:不是,馬凡氏綜合徵是常染色體顯性遺傳,如果夫妻倆決定要孩子的話建議去做基因檢測的機構查一下染色體,如有染色體異常那麼可通過第三代試管嬰兒進行篩查做到優生優育。
常見的染色體疾病彙總
染色體疾病
3、染色體片段缺失
4、染色體平衡易位
5、短QT間期綜合徵
6、長QT間期綜合徵
10、2q37缺失綜合徵
11、SATB2相關綜合徵
12、3q29微缺失綜合徵
13、3q29微重複綜合徵
14、4p綜合徵
15、5q綜合徵
16、5q31.3微缺失綜合症
17、6q24新生兒糖尿病
18、7q11.23重複綜合徵
19、8p11骨髓增殖綜合徵
21、8號染色體重組
22、Kleefstra綜合症
23、12p四體綜合徵
24、14號環形染色體
25、15q13.3微缺失
26、神經母細胞瘤
27、先天性肌營養不良症
28、嗜酸細胞性白血病
29、小兒貓叫綜合徵
30、腦室周圍結節性異位症
31、TAR綜合徵
32、馬凡氏綜合症
33、特納氏綜合症的原因
35、SCS綜合徵
36、膀胱癌基因檢測
37、BWS綜合徵
38、伊曼紐爾綜合症
39、尤文肉瘤
40、視網膜母細胞瘤
41、帕套綜合徵
42、雅克布森綜合症
43、FOXG1綜合症
44、多發性骨髓瘤基因檢測
45、小胖威利綜合徵
46、毛髮鼻指綜合症
47、3p缺失綜合症